All investigational medicinal products (IMPs) go through a series of trials before they can be licensed for use.
There are usually 4 phases of trials in humans (clinical trials). A Phase 1 trial is usually the first time that an IMP is tested in humans and so it will usually investigate the safe dose range and potential side effects, how it is metabolised and whether it might work in patients.
Phase 1 review timelines and 7-day submission
We are working to ensure that the UK routinely delivers ethics opinions for Phase 1 clinical trials within a short timeframe, while maintaining robust ethics review.
All clinical trials of investigational medicinal products (CTIMP) applications, including Phase 1 studies, must use our Combined Review service. This means applicants submit one combined application to the REC and MHRA via the new part of IRAS, and benefit from a more streamlined regulatory and ethics review.
Following feedback from our research community, applicants undertaking Phase 1 clinical trials may reserve a Research Ethics Committee (REC) meeting slot in advance of making their submission and submit an application closer to the REC meeting.
The majority of RECs in the UK that are recognised to review Phase 1 clinical trials in healthy volunteers accept Phase 1 applications submitted up to 7 days before the meeting date.
To make a request for 7-day submission for an application, applicants should contact their preferred REC which is flagged to review Phase 1 clinical trials.
Our fast-track research ethics review is also available for global clinical and phase 1 trials for any disease area.
REC target timelines for fast-track REC applications
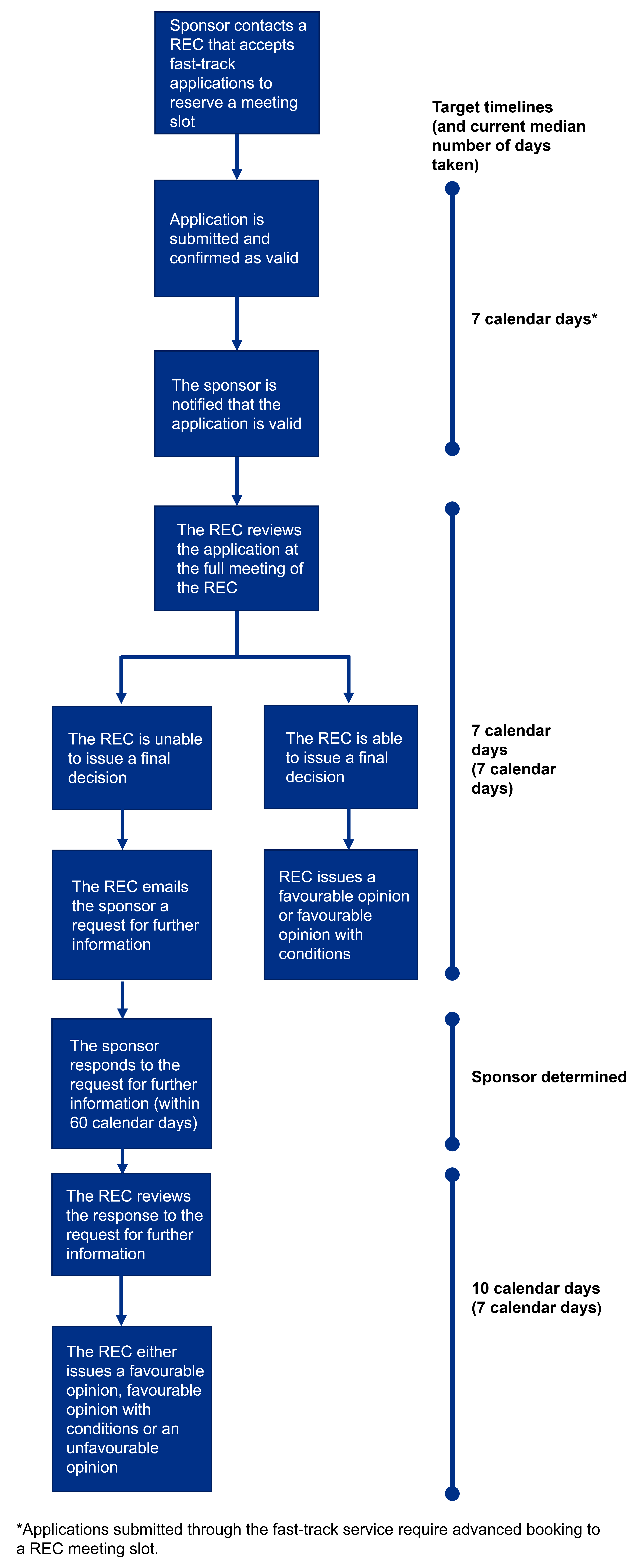
The following diagram outlines the target timeframes that RECs will work towards for all Phase 1 healthy volunteer applications submitted through the fast-track research ethics review service. Please note this diagram does not include the timeframe for Medicines and Healthcare products Regulatory Agency (MHRA) assessment of Phase 1 applications.
The target timelines we have for each part of the process are detailed on the right side of the diagram, in addition to the current average time taken for this to be completed for Phase 1 trial applications (based on performance data for fast-track applications reviewed by RECs between 1 April 2024 and 31 March 2025).
Trial registration and publication of research summaries
If a sponsor submits a Phase 1 clinical trial of investigational medicinal product (CTIMP) application only involving healthy volunteers it will automatically be deferred for all transparency requirements until 30 months after the end of the trial. However the sponsor must still publish a minimal record on a publicly accessible registry (a minimal record can be published with ISRCTN).
This means a sponsor will not be required to include a written request for a deferral as part of the application. It will be applied automatically upon receipt of a Phase 1 CTIMP only involving healthy volunteers. Confirmation that a deferral is in place will be provided as part of the final approval given to a trial.
Deferrals for Phase 1 CTIMPs involving patients
If a trial is a Phase 1 CTIMP involving patients it will not automatically be deferred, even if it will involve both healthy volunteers and patients. Sponsors of these trials will be able to request an initial deferral of the transparency requirements by following the process in our deferrals guidance.
For more information on clinical trial registration and deferrals, visit our research registration page.
The Over-Volunteering Prevention System (TOPS)
The Over-Volunteering Prevention System (TOPS) is a database, free to all UK organisations undertaking Phase 1 trials, that aims to prevent participants from taking part too frequently in trials of new medicines.
Organisations must register participants on TOPS before they are recruited into a clinical trial. This is a condition of the REC favourable opinion in the UK.
Generic Document Review Committee
Clinical trials units often undertake general, non-trial specific advertising and screening procedures to recruit potential trial participants, before inviting them to participate in a specific trial.
These particular activities are not part of the conduct of a trial as defined in the Medicines for Human Use (Clinical Trials) (Amendment) Regulations 2025, meaning there is no legal requirement for the associated documents to be reviewed by a REC. However, it is expected applicants share any proposed generic materials with the REC before they are used in recruitment as a matter of best practice.
To ensure consistency, all generic recruitment materials are reviewed centrally by the Generic Document Review Committee.
This committee is made up of 2 experienced members who are former members of Research Ethics Committees (RECs).
Examples of the types of generic materials reviewed by this group include:
- posters
- website adverts
- scripts for radio broadcasts and television adverts
- website scripts and privacy policies
- patient cards
- participant brochures
- frequently asked questions leaflets
- advertising on social media, for example Facebook or Twitter
- screening information sheets and consent forms
- the HRA template for generic screening information sheets
- GP letters
- Clinical Trial Unit internal participant guidance/rules documentation
- posters and publicity campaigns
To obtain a review of generic materials, please send copies to GDRC@hra.nhs.uk.
Materials submitted to the Generic Document Review Committee should not be related to a particular trial. If documents have been designed for use in a specific Phase 1 clinical trial, they must be submitted to the REC undertaking the ethics review, in line with the Medicines for Human Use (Clinical Trials) (Amendment) Regulations 2025.
Generic Screening Documentation
Our template information sheet and consent form is available for organisations undertaking non-trial specific screening procedures (generic screening) to recruit potential trial participants, before inviting them to take part in a specific clinical trial in the UK.
The generic screening information sheet and consent form template is to be used for the generic screening of healthy volunteers only. It should not be used for enrolling potential participants into a particular trial. There is supporting guidance to provide information on how the generic templates can be used for Phase 1 research.
All trial specific documentation (including Participant Information Sheets and Consent Forms) must be submitted to the Research Ethics Committee and must be ethically approved.
Phase 1 Advisory Group
The Phase 1 Advisory Group was established by the HRA as a forum to discuss issues relating to the ethics review of Phase 1 trials in the UK.
The advisory group brings together:
- HRA operations staff
- Chairs of NHS/Health and Social Care (HSC) RECs with Type 1 recognition to review phase 1 trials
- Phase 1 trials units
- Association of the British Pharmaceutical Industry (ABPI)
- Bio-Industry Association (BIA)
- Contract Clinical Research Association (CCRA)
- Association of Human Pharmacology in the Pharmaceutical Industry (AHPPI)
- Medicines and Healthcare Products Regulatory Agency (MHRA)
- Association of Clinical Research Organizations (ACRO).
The Phase 1 Advisory Group meets twice a year. Topics discussed include operational issues, training for REC members and initiatives to enhance the efficiency and effectiveness of ethics review. If you would like to read minutes of previous meetings, please contact us at foi@hra.nhs.uk
Additional Guidance
MHRA Phase 1 accreditation scheme
The MHRA also publishes guidance for applying for a clinical trial authorisation.
Phase 1 trials: guidelines
- MHRA GCP guide – includes a specific chapter on Phase 1 trials
- GCP forum - a tool to help those involved in CTIMPs comply with the clinical trials legislation and GCP requirements
- ABPI guidelines for Phase 1 Clinical Trials – reflects current EU legislation for the performance of Phase 1 clinical trials
- Strategies to identify and mitigate risks in first in human clinical trials – European Medicines Agency (EMA) guidelines from the Committee for Medicinal Products for Human Use.
Phase 1 trials: ethical issues
- Insurance in Phase 1 Trials guidance developed by the Association for the British Pharmaceutical Industry, the Bio Industry Association and the Clinical Contract Research Association in consultation with the Department of Health and the National Research Ethics Service
- Incentives in Phase 1 Trials – National Research Ethics Advisors’ Panel endorsed guidance regarding payments and incentives in Phase 1 studies, produced by the Phase 1 Advisory Group